High-scoring Review: Origin, Heterogeneity and Targeted Therapy Advances of Cancer-associated Fibroblasts (CAFs)
I. Background Overview
The tumor microenvironment (TME) is a key regulatory hub for tumor development and progression. Signal communication among heterogeneous cells collectively constructs a supportive microenvironment that facilitates cancer cell survival, proliferation, and immune evasion. This concept can be traced back to Paget's "seed and soil" hypothesis. As the most core stromal cells in TME, cancer-associated fibroblasts (CAFs), also known as activated fibroblasts, participate in multiple key aspects of tumor progression through extensive interactions with cancer cells, immune cells, and other stromal cells, including extracellular matrix (ECM) remodeling, immune-suppressive microenvironment construction, and regulation of tumor invasion and metastasis.
Although extensive studies have confirmed the pro-tumor functions of CAFs, some studies have also found that they can act as host defense mechanisms to inhibit tumor progression. This contradiction highlights the high heterogeneity and plasticity of CAFs. In recent years, the development of single-cell RNA sequencing, proteomics, and other technologies has further resolved the composition of CAF subpopulations based on their unique transcriptional characteristics, confirming that stromal myofibroblast populations undergo dynamic heterogeneous changes depending on environmental contexts, breaking the traditional perception of CAFs as a single cell type. However, the precise origin of CAFs remains unclear, the regulatory mechanisms of their heterogeneity still need in-depth analysis, and previous pan-targeted therapies against CAFs have mostly ended in clinical failure, even accelerating tumor progression.
Therefore, this article systematically reviews the origin and heterogeneity characteristics of CAFs, analyzes the regulatory mechanisms of their phenotypic and functional diversity, explores therapeutic strategies for precise targeting of pro-tumor CAFs, and summarizes the latest research progress on specific targeting of pro-tumor CAFs and inhibition of their activity, providing potential strategies for cancer treatment.
II. Origin Diversity of CAFs
CAFs are not a single cell population; their origin is highly diverse, mainly including two categories: local intrinsic cell sources and bone marrow-recruited cell sources. CAFs from different origins exhibit significant differences in phenotype and function.
Local intrinsic cell sources: This is the most direct source of CAFs, mainly including tissue-resident fibroblasts and quiescent stellate cells. CAFs in the pancreas and liver are traditionally considered to originate from pancreatic stellate cells (PSCs) and hepatic stellate cells (HSCs), respectively. Under stimulation by chemokines, cytokines, and microRNAs secreted by tumor cells, PSCs and HSCs are activated, acquiring myofibroblast-like features and CAF transcriptional characteristics. They maintain their activity through autocrine loops and participate in tumor stromal hyperplasia. Notably, in vivo lineage tracing studies have found that a subset of CAFs in breast cancer can originate from adipocytes, and the conversion process is regulated by the TGF-β/SMAD signaling pathway.
Bone marrow-recruited cell sources: Although CAFs mainly originate locally, lineage tracing studies in mouse models and human samples have confirmed that bone marrow cells can also participate in CAF pool formation, involving various tumors such as colorectal adenoma, gastric cancer, liver cancer, pancreatic cancer, and breast cancer. Bone marrow-derived mesenchymal stem cells (MSCs) can differentiate into CAF subpopulations under the regulation of TGF-β, WNT, and IL-6/STAT3 signals secreted by tumor cells. In addition, bone marrow-derived macrophages/monocytes can differentiate into CAFs through macrophage-to-fibroblast transition (MFT) under specific conditions, and this process is related to the expression of transcription factors such as Twist1 and Snail.
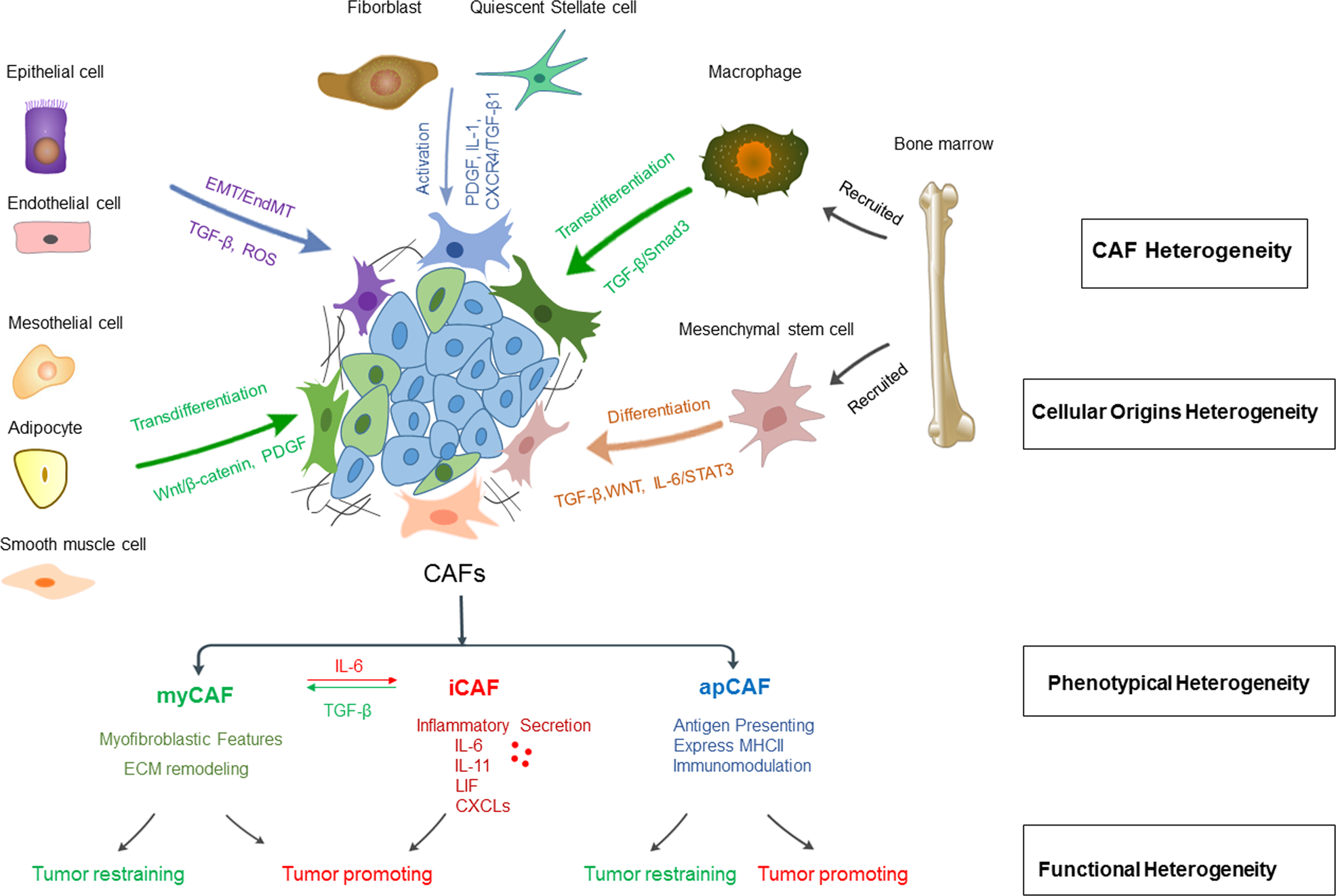
III. Phenotypic and Functional Heterogeneity of CAFs
The heterogeneity of CAFs is the core reason for their functional contradictions, mainly reflected in three aspects: phenotypic markers, spatial distribution, and functional activity. Functional classification is based on studies of pancreatic cancer organoids co-cultured with mouse PSCs, and has gradually expanded to multiple tumor types.
1. Classical CAF subpopulations: Currently identified core subpopulations include three types, each with unique markers and functions:
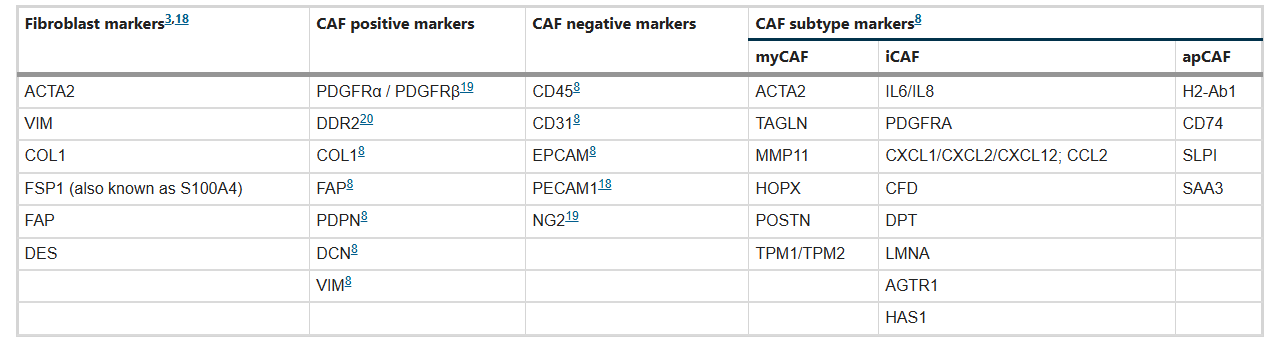
○ Myofibroblast-like CAFs (myCAFs): Highly express α-smooth muscle actin (α-SMA), ACTA2, TAGLN, and other markers, low expression of IL-6. Activated through direct contact with tumor cells, located around tumor foci. Characterized by enhanced contractile ability and massive ECM synthesis. Their functions are dualistic—on one hand, they promote tumor progression by enhancing ECM stiffness and promoting tumor cell invasion; on the other hand, they may inhibit tumor growth by restricting tumor cell movement and limiting nutrient supply in some contexts.
○ Inflammatory CAFs (iCAFs): Low expression of α-SMA, high expression of IL-6, IL-8, CXCL family, and other inflammatory factors. Induced by IL-1α, TNFα, etc., secreted by tumor cells. Located in areas away from tumor cells. Mainly promote tumor cell proliferation, metastasis, and chemotherapy resistance through secretion of inflammatory cytokines and growth factors, with clear pro-tumor functions.
○ Antigen-presenting CAFs (apCAFs): Characterized by expression of major histocompatibility complex class II molecules (MHCII), markers include H2-Ab1, CD74, etc. Possess immunomodulatory functions, with context-dependent effects—in breast cancer and pancreatic cancer, they can induce regulatory T cell proliferation and promote immune suppression; however, in some tumor types, they may enhance anti-tumor immune responses by presenting tumor antigens.
2. Other emerging CAF subpopulations: With the application of single-cell sequencing technology, new CAF subpopulations have been successively discovered in different tumors, such as vascular-associated CAFs (vCAFs), circulating CAFs (cCAFs), developmental CAFs (dCAFs) in breast cancer, and CAFs in cholangiocarcinoma that respond to placental growth factor (PlGF). These emerging subpopulations further enrich the understanding of CAF heterogeneity and provide new targets for tumor treatment.
3. Plasticity of CAFs: CAF subpopulations are not permanent terminally differentiated states but possess high plasticity. They can transform between subpopulations through regulation of specific signaling pathways. TGF-β signaling can induce iCAFs to transform into myCAFs, while IL-6 signaling can promote myCAFs to transform into iCAFs. This plasticity enables CAFs to dynamically adapt to changes in the tumor microenvironment and participate in different stages of tumor progression.
Table: Pan-CAF markers and specific markers for CAF subtype identification
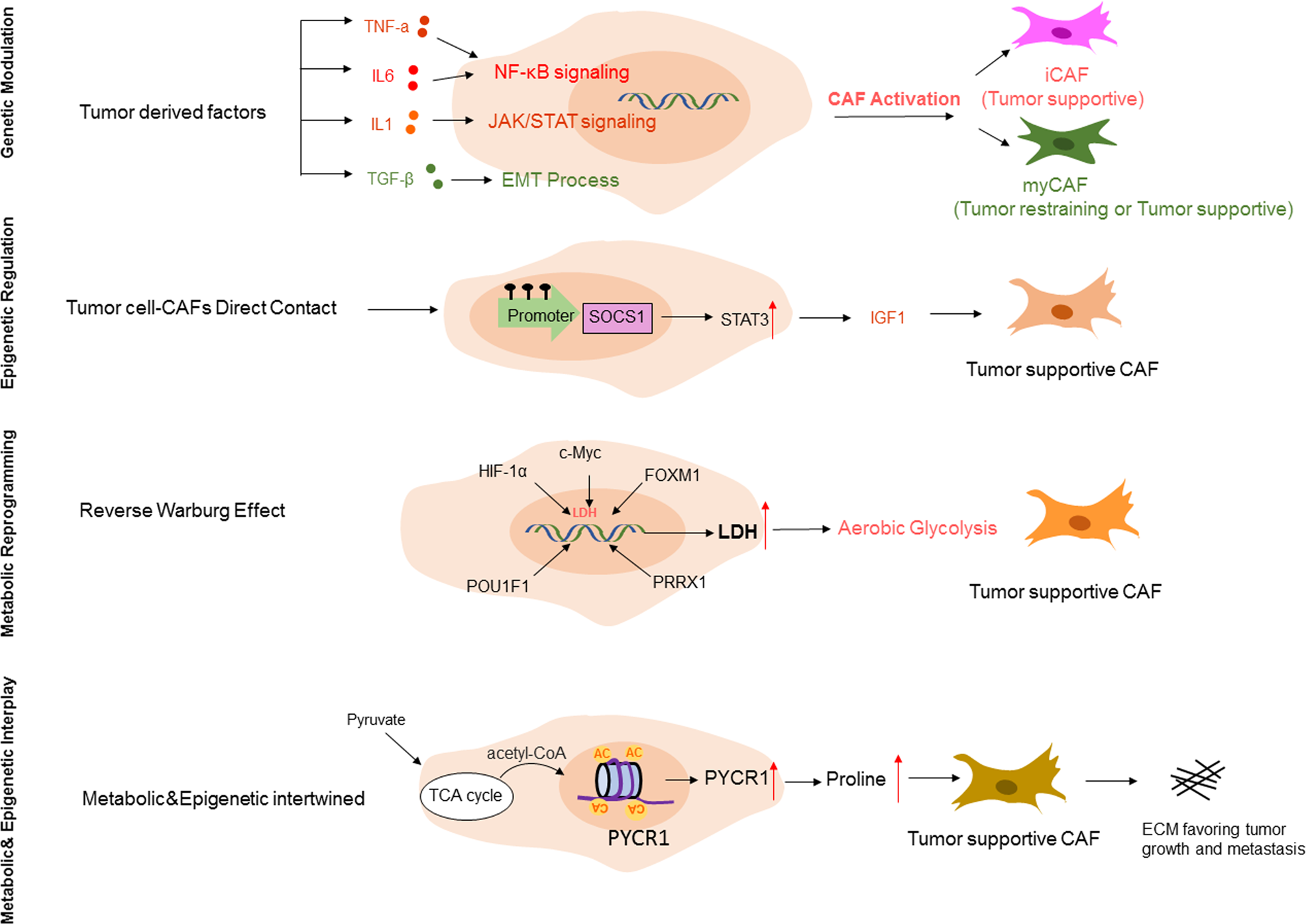
IV. Regulatory Mechanisms of CAF Heterogeneity
The heterogeneity of CAFs is mainly regulated by tumor cell-induced genetic regulation, epigenetic modifications, and metabolic reprogramming, which synergistically shape the phenotypic and functional diversity of CAFs.
1. Genetic regulation: Tumor cells secrete growth factors (TGF-β, EGF, PDGF, etc.) and cytokines (IL-6, IL-1β, etc.) to activate key signaling pathways in CAFs, thereby regulating CAF subpopulation differentiation. For example, TGF-β can induce the transformation of fibroblasts into myCAFs through the SMAD pathway; IL-1α can activate NF-κB signaling to promote the formation of iCAFs. In addition, tumor cells can also regulate CAF function through direct cell-cell contact, such as through Notch and Hedgehog signaling pathways.
2. Epigenetic modifications: Although CAFs rarely undergo genetic abnormalities, they undergo significant epigenomic changes in the TME, mainly including DNA methylation and histone modifications. Abnormal DNA methylation (local hypermethylation, global hypomethylation) can regulate the ECM remodeling ability and immunomodulatory functions of CAFs; histone modifications (such as H3 acetylation) can regulate the expression of proline synthesis key enzyme (PYCR1), promoting the pro-tumor function of CAFs. In addition, non-coding RNAs (miRNAs, lncRNAs) also play an important role in the regulation of CAF heterogeneity.
3. Metabolic reprogramming: CAF activation is accompanied by metabolic phenotype transformation, mainly manifested as the "reverse Warburg effect"—CAFs produce energy metabolites such as lactate through aerobic glycolysis, providing energy for tumor cell oxidative phosphorylation while acquiring pro-tumor functions. Transcription factors such as hypoxia-inducible factor (HIF-1α), c-Myc, and FOXM1 can regulate glycolysis-related enzymes in CAFs, promoting metabolic reprogramming and CAF activation. In addition, lipid metabolism and amino acid metabolism also participate in the regulation of CAF heterogeneity.
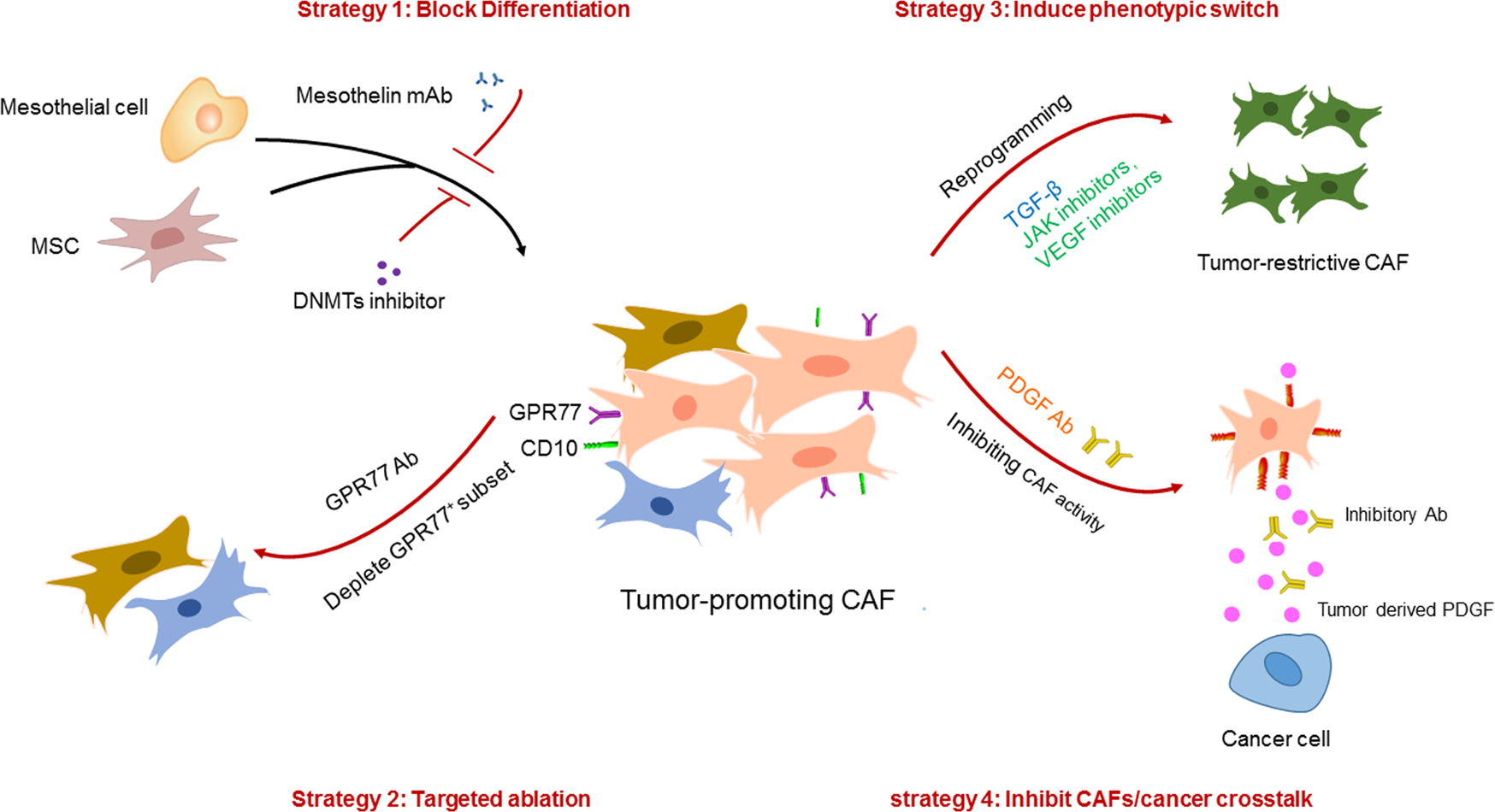
V. Anticancer Therapeutic Strategies Targeting CAFs
1. Inhibit Precursor Cell Differentiation into Pro-tumor CAFs
The core is to block the activation and differentiation of CAF precursor cells, reducing the production of pro-tumor CAFs from the source, mainly including two directions:
Target precursor cell activation signals: Targeting precursor cell activation signals induced by tumor cells, such as IL-1, TGF-β, etc. However, due to their pleiotropic effects, direct targeting of these signaling molecules has limitations. Therefore, precursor cell-specific markers can be targeted, such as the mesothelial cell marker mesothelin (MSLN). Its monoclonal antibody can effectively block mesothelial cell activation and apCAF generation, reducing immune suppression.
Epigenetic reprogramming: Using DNA methyltransferase (DNMT) inhibitors (such as decitabine), NNMT inhibitors, etc., to reverse epigenetic abnormalities in CAFs and inhibit their differentiation into pro-tumor phenotypes. For example, decitabine pretreatment can prolong tumor-free survival in pancreatic cancer xenograft models, and NNMT inhibitors can reverse the CAF phenotype in ovarian cancer to restore the normal omental fibroblast phenotype.
2. Selective Ablation of Pro-tumor CAF Subpopulations
Based on specific markers of CAF subpopulations, precise ablation of pro-tumor CAFs is achieved while avoiding damage to tumor-suppressive CAFs, mainly including:
Target CAF subpopulation-specific markers: The CD10+GPR77+ CAF subpopulation is associated with cancer stem cell maintenance and chemotherapy resistance. Targeting this subpopulation (e.g., GPR77 neutralizing antibody) can inhibit tumor growth and reverse chemotherapy resistance. In addition, targeting HSCs, the main source of CAFs in cholangiocarcinoma, through Cre-mediated diphtheria toxin (DT) treatment to specifically ablate HSCs can reduce CAF accumulation and inhibit tumor progression.
Target CAF activation signaling pathways: Design multifunctional targeting molecules to simultaneously block CAF activation signals and tumor cell interactions. For example, Ate-Grab (a PlGF/VEGF decoy receptor targeting PD-L1) can specifically bind to PD-L1 in tumor tissue, while chelating PlGF/VEGF to inhibit CAF activation, alleviate chemotherapy-induced stromal hyperplasia in pancreatic cancer, and improve chemotherapy efficacy.
3. Induce CAF Phenotype Transformation towards Tumor-Suppressive Direction
Utilizing the plasticity of CAFs, by regulating key signaling pathways, transforming pro-tumor CAF subpopulations into tumor-suppressive phenotypes has higher feasibility and safety, mainly including:
Regulate TGF-β/IL-6 signaling pathways: TGF-β signaling can induce iCAFs to transform into myCAFs, reducing inflammatory factor secretion and alleviating immune suppression. JAK inhibitors can block the IL-1α/JAK/STAT signaling pathway, transforming iCAFs into myCAFs and inhibiting pancreatic cancer progression. In addition, TGF-β1 overexpression can promote the transformation of CAFs into a tumor-suppressive phenotype, inhibiting tumor angiogenesis.
Target CAF function-related molecules: Targeting CD16+ CAFs (associated with trastuzumab resistance), targeting their key activation molecule VAV2 can block their activation, reverse stromal hyperplasia, and restore trastuzumab sensitivity. IL-1 receptor antagonist (anakinra) can inhibit iCAF formation and is currently in phase I clinical trials for pancreatic cancer, combined with standard chemotherapy.
4. Inhibit Pro-tumor Activity of Mature CAFs
By blocking the interaction between CAFs and tumor cells, inhibiting the pro-tumor functions of CAFs without changing the CAF phenotype, specifically including:
Block paracrine signals between CAFs and tumor cells: PDGF ligands expressed by tumor cells bind to PDGF receptors expressed by CAFs, promoting breast cancer transformation to basal-like subtypes. Targeting the PDGF pathway can transform tumors into endocrine therapy-sensitive luminal subtypes. In pancreatic cancer, p53 mutations induce CAFs to secrete heparan sulfate proteoglycan 2 (HSPG2), promoting tumor cell stemness; targeting HSPG2 can inhibit tumor progression.
Target ECM remodeling function of CAFs: ECM components secreted by CAFs (such as collagen, hyaluronic acid) can form physical barriers to promote tumor progression. Targeting ECM synthesis-related molecules, such as PYCR1, can reduce ECM deposition, break the tumor stromal barrier, and improve the delivery efficiency of therapeutic drugs.
VI. Conclusion
The core contribution of this review article lies in systematically sorting out the origin diversity, phenotypic and functional heterogeneity of CAFs, and their regulatory mechanisms. It proposes four categories of precise therapeutic strategies targeting pro-tumor CAFs, and integrates CAF-related biomarkers and clinical research progress. Through clear diagrams, it visually presents a complete research system of "Origin - Heterogeneity - Regulatory Mechanisms - Therapeutic Strategies", which has important research value and academic contributions.
In terms of research value, this literature systematically integrates the multi-source origins of CAFs for the first time, clarifies the core association between phenotypic heterogeneity and functional contradictions, and fills the gaps in existing research where CAF regulatory mechanisms are not systematically classified and therapeutic strategies are not closely combined with heterogeneity. The proposed strategies such as epigenetic reprogramming, CAF phenotype transformation, and subpopulation-specific ablation have strong clinical translation potential, providing clear directions for the development of next-generation stroma-targeted therapies. The organized CAF-related biomarker system provides important references for CAF subpopulation identification, efficacy prediction, and prognosis assessment, promoting CAF-targeted therapy from "pan-targeting" to "precision targeting".
At the same time, this literature also points out the limitations of current research: The functional classification of CAF subpopulations still needs further improvement, and the hierarchical relationships and transformation mechanisms between different subpopulations have not been fully clarified; the clinical application of CAF-specific markers still has limitations, lacking precise markers that can be widely used clinically; the clinical translation of CAF-targeted therapy still faces many challenges, requiring more rigorous preclinical studies to clarify the applicable scenarios of therapeutic strategies. Future research needs to combine single-cell sequencing, lineage tracing, and other technologies to deeply analyze the heterogeneity and plasticity of CAFs, develop more precise targeting strategies, and promote the clinical application of CAF-targeted therapy to provide new breakthroughs for cancer treatment.
References
Yang D, Liu J, Qian H, Zhuang Q. Cancer-associated fibroblasts: from basic science to anticancer therapy. Exp Mol Med. 2023 Jul;55(7):1322-1332. doi: 10.1038/s12276-023-01013-0. Epub 2023 Jul 3. PMID: 37394578; PMCID: PMC10394065.