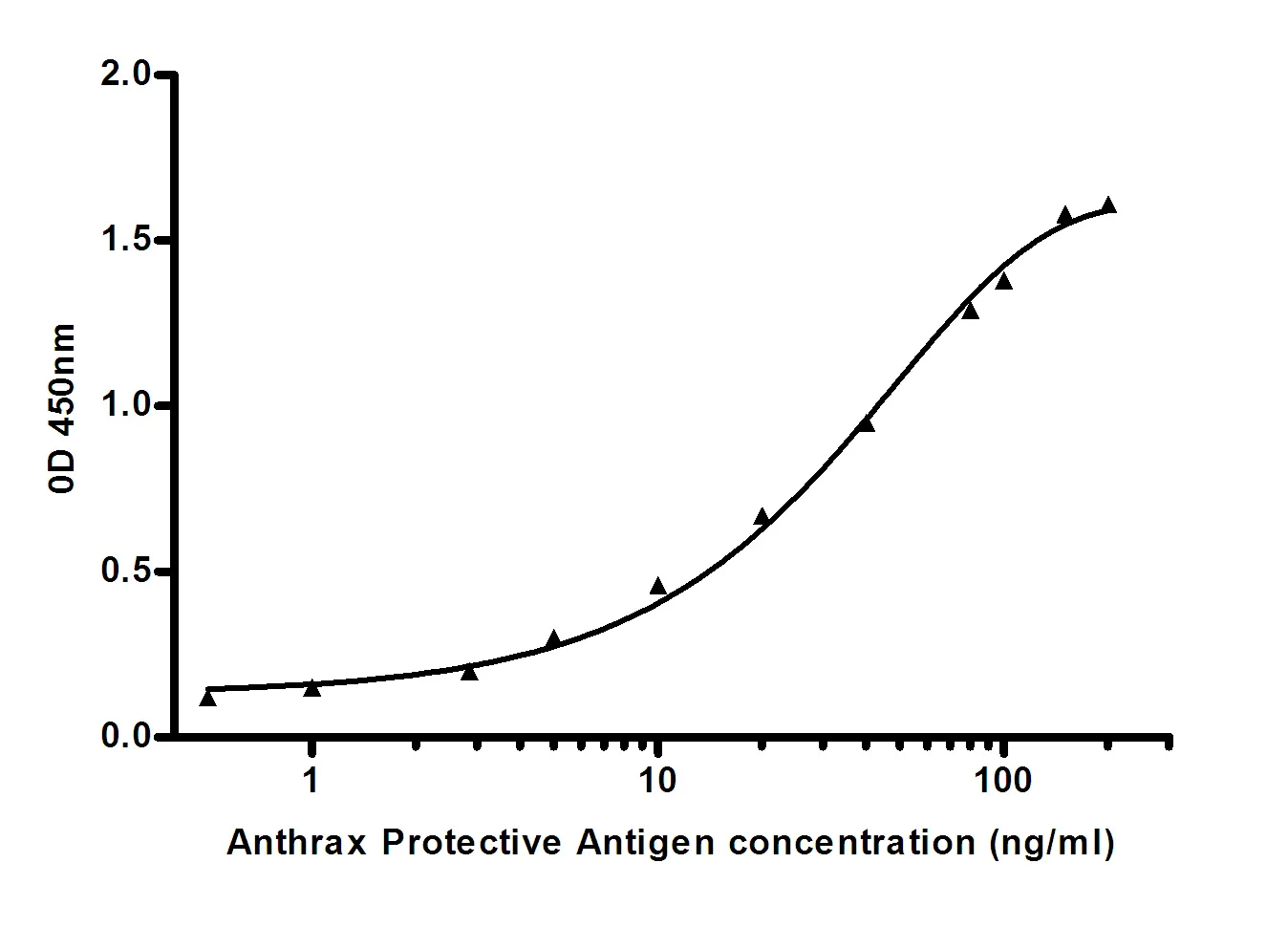
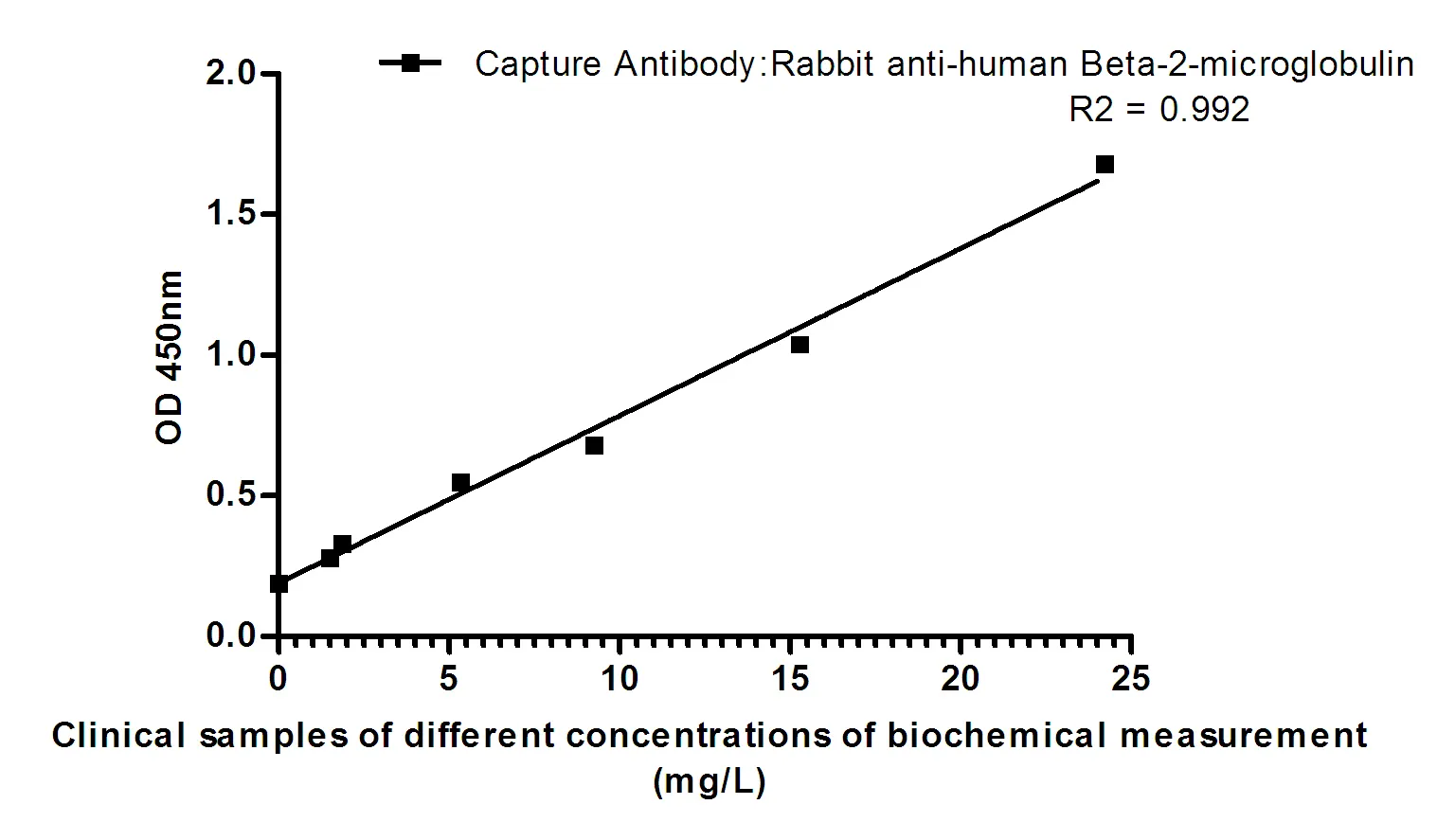
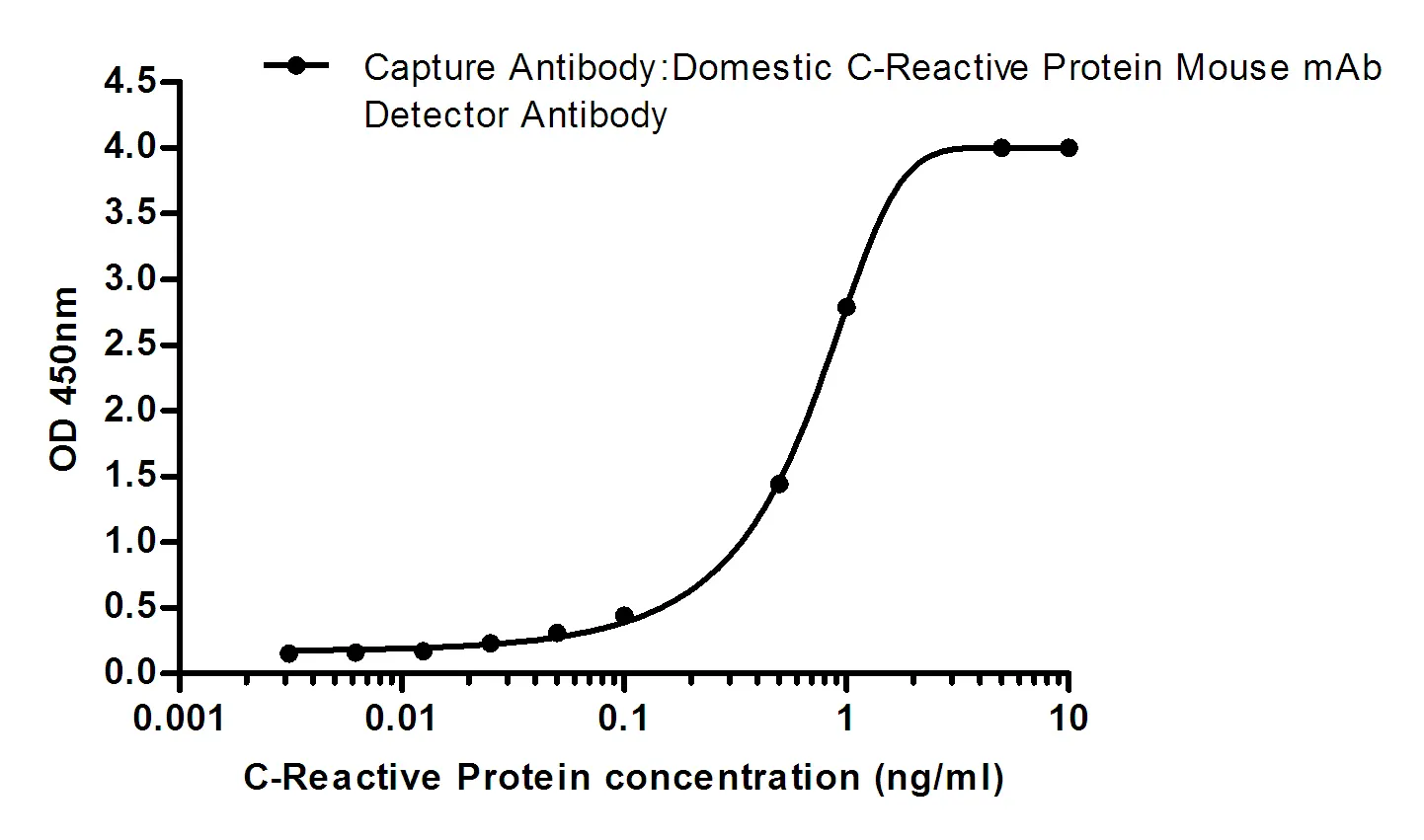
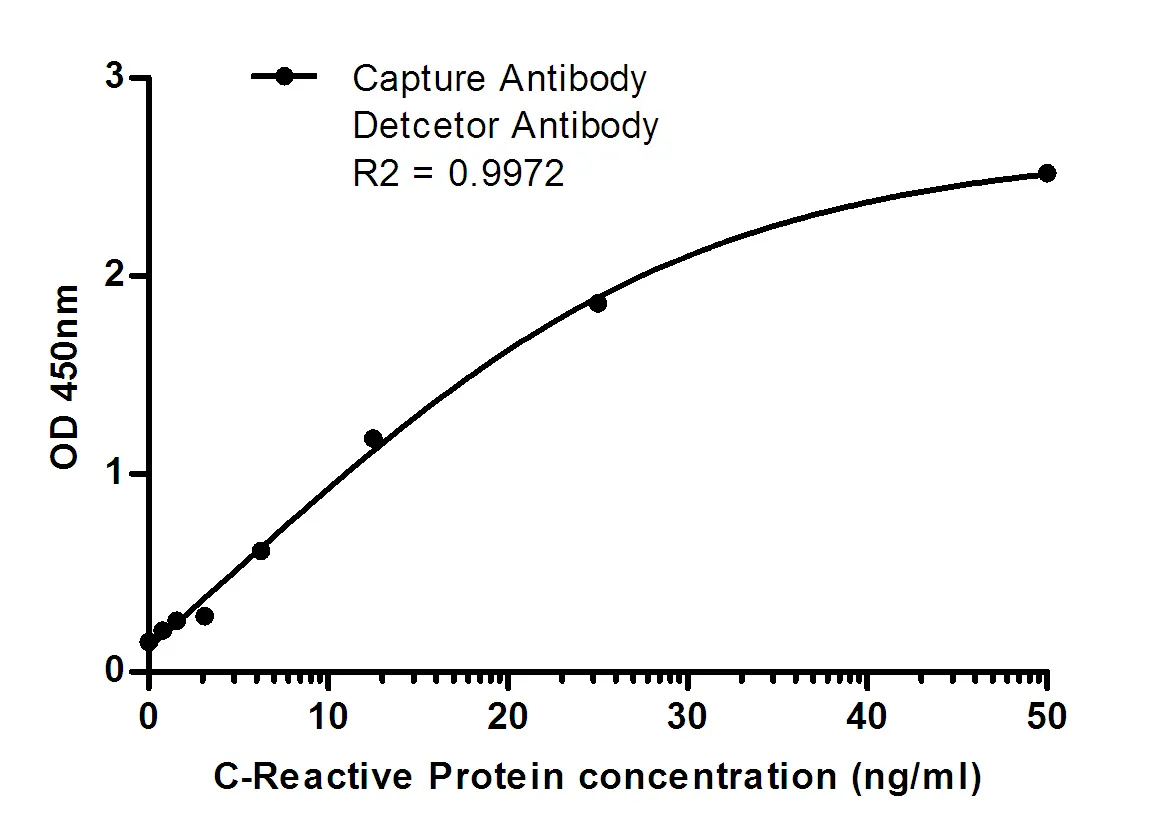
Summary
Performance
Immunogen
Application
Background
The protein encoded by this gene belongs to the superfamily of small heat-shock proteins containing a conservative alpha-crystallin domain at the C-terminal part of the molecule. The expression of this gene in induced by estrogen in estrogen receptor-positive breast cancer cells, and this protein also functions as a chaperone in association with Bag3, a stimulator of macroautophagy. Thus, this gene appears to be involved in regulation of cell proliferation, apoptosis, and carcinogenesis, and mutations in this gene have been associated with different neuromuscular diseases, including Charcot-Marie-Tooth disease. [provided by RefSeq, Jul 2008],caution:Was reported (PubMed:10833516) to have a protein kinase activity and to act as a Mn(2+)-dependent serine-threonine-specific protein kinase.,disease:Defects in HSPB8 are the cause of Charcot-Marie-Tooth disease type 2L (CMT2L) [MIM:608673]. CMT2L is an axonal form of Charcot-Marie-Tooth disease. Axonal CMT neuropathies are characterized by signs of axonal regeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy.,disease:Defects in HSPB8 are the cause of distal hereditary motor neuronopathy type 2A (HMN2A) [MIM:158590]; also known as distal hereditary motor neuropathy type IIA or spinal Charcot-Marie-Tooth disease IIA. Distal hereditary motor neuronopathies constitute a heterogeneous group of neuromuscular disorders caused by selective impairment of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs.,function:Displays temperature-dependent chaperone activity.,induction:By 17-beta-estradiol.,PTM:Phosphorylated.,similarity:Belongs to the small heat shock protein (HSP20) family.,subunit:Monomer. Interacts with HSPB1.,tissue specificity:Predominantly expressed in skeletal muscle and heart.,
Research Area
Signal Transduction